
Uncovering the Link to Combating Muscle Atrophy Caused by Aging and Immobility
Researchers elucidate key underlying mechanisms and outline potential avenues towards new treatments
Muscle atrophy is a prevalent condition in today’s societies, but many of the roles that mitochondria play in the process remain unclear. In a recent study, researchers from Fujita Health University investigated how muscle atrophy relates to the tethering of mitochondria to the endoplasmic reticulum and how the Notch signaling pathway is involved. Their findings reveal some of the mechanisms behind muscle atrophy and could pave the way for new treatments.
The loss of muscle mass, or muscle atrophy, is a relatively common condition in today’s aging and increasingly sedentary societies. While the disuse of muscles is the most frequent catalyst for muscle atrophy, there are several other possible causes, including chronic diseases, injury, and exposure to low-gravity environments, such as in spaceships. Despite being a prevalent condition, its underlying mechanisms are complex and not entirely understood.
Scientists have shown that mitochondria play essential roles during muscle development, regeneration, and maintenance. In particular, muscle stem cells and differentiated muscle fibers (myofibers) need a lot of energy to become fully mature and functional. Thus, problems with mitochondria can immediately translate to muscle diseases, including muscle atrophy.
In a recent study, a team of researchers including Junior Associate Professor Takahiko Sato from Fujita Health University, Japan, revealed the close relationship that exists between muscle atrophy, development, and regeneration and the tethering of mitochondria to the endoplasmic reticulum (ER). Their findings were recently published in eLife on December 15, 2023.
In healthy cells, there are regions in the ER called mitochondria-associated membranes (MAMs) that can be reversibly tethered to the mitochondria. This anchoring serves many functions, such as calcium homeostasis (balance) and regulation of metabolism and mitochondrial morphology. However, whether and how MAMs are involved in the context of muscle atrophy is unclear.
Motivated by this knowledge gap, the research team conducted a variety of experiments involving ‘Mitofusin2’ (MFN2), a protein that is necessary for mitochondria tethering in MAMs. Skeletal muscle cells cultured in a microgravity environment, known to result in muscle atrophy, exhibited a sharp reduction in the number of MAMs, as well as lower levels of MFN2, alongside typical symptoms of muscle atrophy. Likewise, human cells with a mutated MFN2 gene exhibited similar traits, as well as abnormalities in mitochondrial fission and lower energy (or ATP) production, indicating problems in the generation of ‘cellular energy’.
A closer look at the gene expression profiles in these atrophied muscle cells showed that they all exhibited an upregulation in the Notch signaling pathway. This pathway is essential for cell communication and regulates multiple cell processes, including cell proliferation, differentiation, and programmed death. Using the gammasecretase inhibitor DAPT, a known suppressor of Notch signaling, the researchers managed to revert the effects of MFN2 deficiency and partially restore mitochondrial morphology and function, as well as the number of MAMs.
The team further investigated the relationship between MAMs, MFN2, and Notch signaling in muscle atrophy in mice. They analyzed how MFN2 deficiency induced in muscle tissue and the inhibition of Notch signaling affected muscle regeneration, after either repeated injury or transplantation of muscle stem cells. The results were consistent with the in vitro experiments, as Dr. Sato remarks, “Our tests demonstrate that the restoration of ER-mitochondria contacts through the regulation of Notch signaling is sufficient to partially alleviate the bioenergetic defects in atrophied muscle cells. This suggests that treatment with gamma-secretase inhibitors may be a viable therapeutic option in pathological conditions in which MFN2 is involved.” In other words, the findings may reveal new avenues towards the treatment of skeletal muscle atrophy caused by mitochondrial abnormalities.
Further research efforts will be needed to get a more comprehensive understanding of how the complex orchestration of mitochondria and the associated signaling pathways are related to the loss of muscle mass. With eyes on the future, Dr. Sato remains optimistic, as he concludes, “In the future, we may be able to develop new therapeutic treatments that prevent or ameliorate muscle atrophy caused by aging and immobility.”
Let us hope he is correct so that more people can enjoy healthy and happy lives!
Image: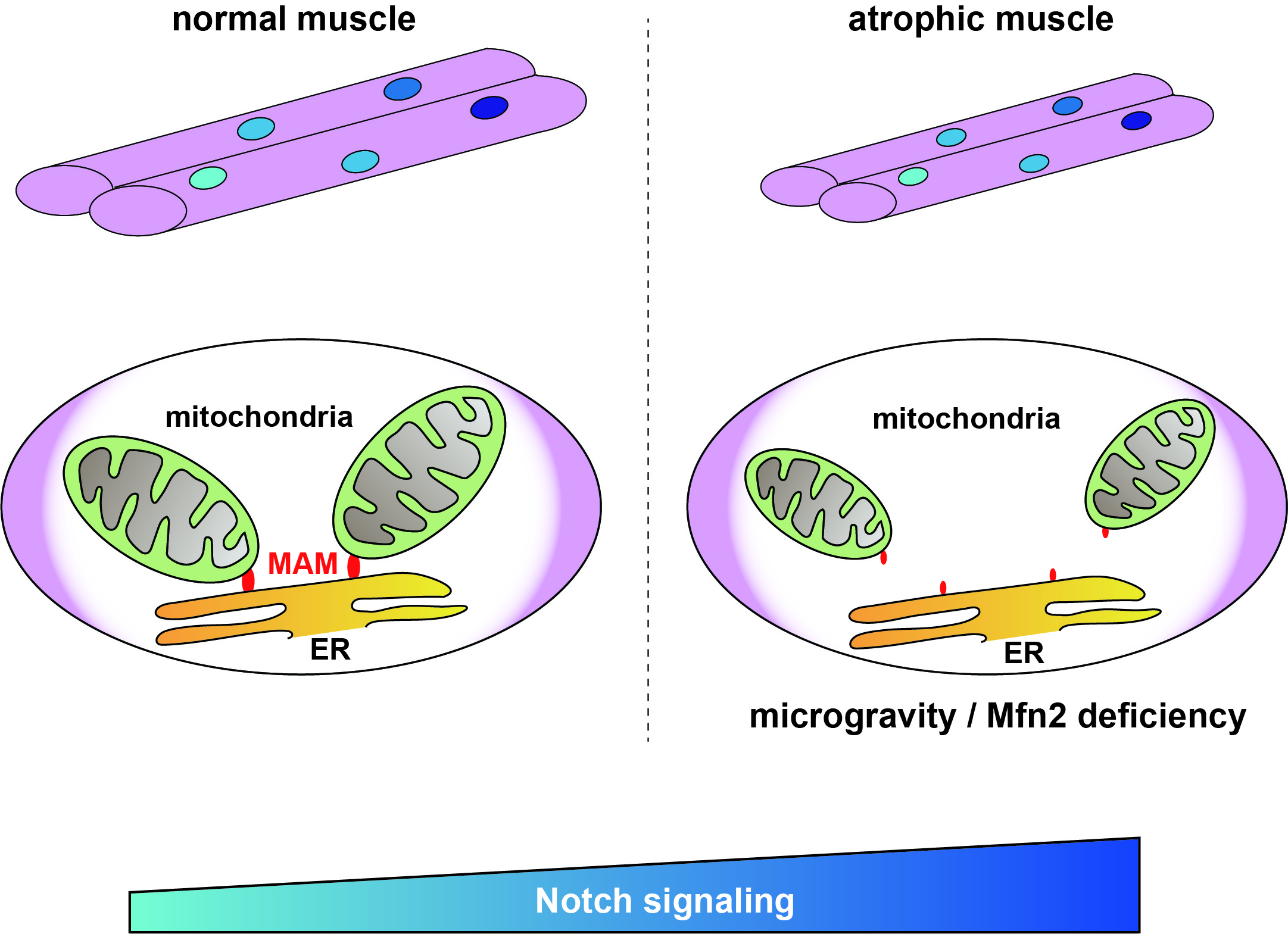 Image title: MAM and Notch signal expression levels during muscle atrophy
Image title: MAM and Notch signal expression levels during muscle atrophy
Image caption: Mitochondrial morphology in myotubes and muscle stem cells coordinate to control Notch signaling via MAM.
Image credit: Takahiko Sato from Fujita Health University
License type: Original content
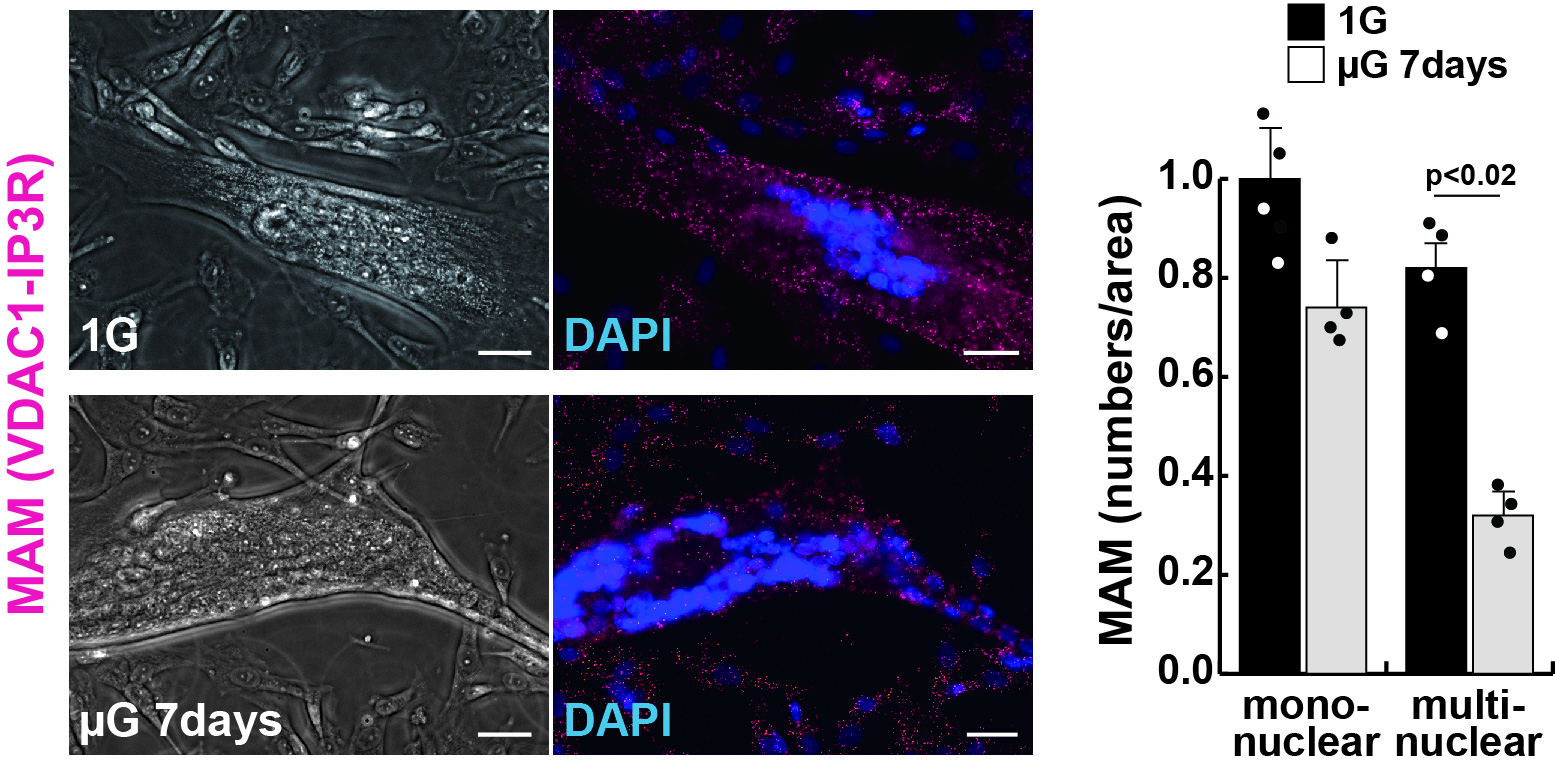
Image title: Mitochondria-associated membrane (MAM) number decreases in human muscle cells cultured under microgravity
Image caption: MAM (red) decreases in human muscle cells cultured in microgravity for 7 days (µG 7days). The MAM number per unit area was significantly decreased differentiated multinucleate myotubes, but not in undifferentiated mononuclear myoblasts.
Image credit: Takahiko Sato from Fujita Health University
Image source: Adapted from here
License type: Original content
Muscle atrophy is a prevalent condition in today’s societies, but many of the roles that mitochondria play in the process remain unclear. In a recent study, researchers from Fujita Health University investigated how muscle atrophy relates to the tethering of mitochondria to the endoplasmic reticulum and how the Notch signaling pathway is involved. Their findings reveal some of the mechanisms behind muscle atrophy and could pave the way for new treatments.
The loss of muscle mass, or muscle atrophy, is a relatively common condition in today’s aging and increasingly sedentary societies. While the disuse of muscles is the most frequent catalyst for muscle atrophy, there are several other possible causes, including chronic diseases, injury, and exposure to low-gravity environments, such as in spaceships. Despite being a prevalent condition, its underlying mechanisms are complex and not entirely understood.
Scientists have shown that mitochondria play essential roles during muscle development, regeneration, and maintenance. In particular, muscle stem cells and differentiated muscle fibers (myofibers) need a lot of energy to become fully mature and functional. Thus, problems with mitochondria can immediately translate to muscle diseases, including muscle atrophy.
In a recent study, a team of researchers including Junior Associate Professor Takahiko Sato from Fujita Health University, Japan, revealed the close relationship that exists between muscle atrophy, development, and regeneration and the tethering of mitochondria to the endoplasmic reticulum (ER). Their findings were recently published in eLife on December 15, 2023.
In healthy cells, there are regions in the ER called mitochondria-associated membranes (MAMs) that can be reversibly tethered to the mitochondria. This anchoring serves many functions, such as calcium homeostasis (balance) and regulation of metabolism and mitochondrial morphology. However, whether and how MAMs are involved in the context of muscle atrophy is unclear.
Motivated by this knowledge gap, the research team conducted a variety of experiments involving ‘Mitofusin2’ (MFN2), a protein that is necessary for mitochondria tethering in MAMs. Skeletal muscle cells cultured in a microgravity environment, known to result in muscle atrophy, exhibited a sharp reduction in the number of MAMs, as well as lower levels of MFN2, alongside typical symptoms of muscle atrophy. Likewise, human cells with a mutated MFN2 gene exhibited similar traits, as well as abnormalities in mitochondrial fission and lower energy (or ATP) production, indicating problems in the generation of ‘cellular energy’.
A closer look at the gene expression profiles in these atrophied muscle cells showed that they all exhibited an upregulation in the Notch signaling pathway. This pathway is essential for cell communication and regulates multiple cell processes, including cell proliferation, differentiation, and programmed death. Using the gammasecretase inhibitor DAPT, a known suppressor of Notch signaling, the researchers managed to revert the effects of MFN2 deficiency and partially restore mitochondrial morphology and function, as well as the number of MAMs.
The team further investigated the relationship between MAMs, MFN2, and Notch signaling in muscle atrophy in mice. They analyzed how MFN2 deficiency induced in muscle tissue and the inhibition of Notch signaling affected muscle regeneration, after either repeated injury or transplantation of muscle stem cells. The results were consistent with the in vitro experiments, as Dr. Sato remarks, “Our tests demonstrate that the restoration of ER-mitochondria contacts through the regulation of Notch signaling is sufficient to partially alleviate the bioenergetic defects in atrophied muscle cells. This suggests that treatment with gamma-secretase inhibitors may be a viable therapeutic option in pathological conditions in which MFN2 is involved.” In other words, the findings may reveal new avenues towards the treatment of skeletal muscle atrophy caused by mitochondrial abnormalities.
Further research efforts will be needed to get a more comprehensive understanding of how the complex orchestration of mitochondria and the associated signaling pathways are related to the loss of muscle mass. With eyes on the future, Dr. Sato remains optimistic, as he concludes, “In the future, we may be able to develop new therapeutic treatments that prevent or ameliorate muscle atrophy caused by aging and immobility.”
Let us hope he is correct so that more people can enjoy healthy and happy lives!
Image:
Image caption: Mitochondrial morphology in myotubes and muscle stem cells coordinate to control Notch signaling via MAM.
Image credit: Takahiko Sato from Fujita Health University
License type: Original content
Image title: Mitochondria-associated membrane (MAM) number decreases in human muscle cells cultured under microgravity
Image caption: MAM (red) decreases in human muscle cells cultured in microgravity for 7 days (µG 7days). The MAM number per unit area was significantly decreased differentiated multinucleate myotubes, but not in undifferentiated mononuclear myoblasts.
Image credit: Takahiko Sato from Fujita Health University
Image source: Adapted from here
License type: Original content
Reference
Title of original paper
The reciprocal regulation between mitochondrial-associated membranes and Notch signaling in skeletal muscle atrophyJournal
eLifeDOI
10.7554/eLife.89381About Junior Associate Professor Takahiko Sato
Dr. Takahiko Sato conducts basic research using skeletal muscle cells, which are essential precursors in muscle development and regeneration. He aims to establish new cell culture technologies using stem muscle cells, with the ultimate goal of treating challenging muscle diseases and reverting the deteriorating effects of aging on muscles. He has published over 26 scientific articles on these topics.
Funding information
This work was supported by the Japan Society for the Promotion of Science, KAKENHI grants 17K01859, 18H04061, 23K10971, Japan Agency for Medical Research and Development (AMED) CREST 21gm0910009h0506, JP16gm0810009, the Nakatomi Memorial Foundation, and the Hori Sciences and Arts Foundation.