![Institute for Comprehensive Medical Science [ICMS], Fujita Health University, Division of Gene Expression Mechanism](image/etoptitle.png)


Research
Search for the mechanisms of aberrant splicing mediated by oncogene product HMGA1
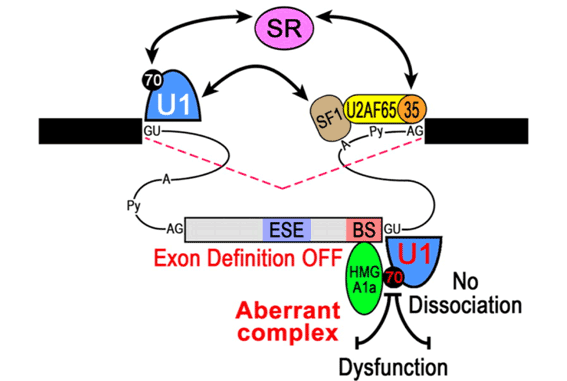
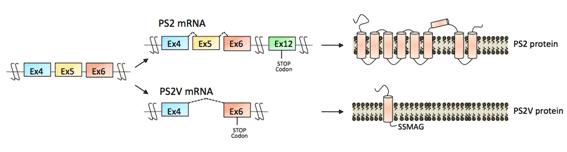
Most of the senile cases of the Alzheimer’s disease (AD) have no mutation in the known AD-associated four genes and thus it is not familial but sporadic. In such sporadic AD, however, the specific aberrant splicing (skipping of exon 5) was observed at high frequency in presenilin-2 (PS2) gene transcript, which is one of the known Alzheimer’s disease (AD)-associated genes. This aberrant splicing generates the truncated deleterious protein PS2V and it accumulates as visible PS2V bodies in the brains of sporadic AD patients (see Figure), which is one of the risk factors of neuronal cell death in sporadic AD. We found that HMGA1a, known as an oncogene product, binds to a specific HMGA1a-binding site adjacent to the 5’ splice site of the PS2 pre-mRNA, and it causes aberrant skipping of exon 5. We eventually elucidated the detail molecular mechanism of this process. We are going to analyze the structure of the HMGA1–U1 snRNP–RNA complex, which plays a central role in the mechanism. We are searching for another target genes that potentially cause splicing defects leading to diseases.
Analysis of schizophrenia from the linked aberrant splicing events
Schizophrenia is a very common (~1% of the population is affected) but still mysterious mental disorder, caused by complex interaction of both genetic and environmental factors. There have been accumulating reports of aberrant splicing events in the related genes in schizophrenia patients, however, not much study has been carried out with regard to a splicing abnormality implicated in this disease. Interestingly, the aberrant splicing of PS2 found in sporadic AD (see above) was also observed in the brains of bipolar disorder and schizophrenia. We detected markedly higher HMGA1a mRNA and the increased HMGA1a protein in the lymphocytes of schizophrenia patients. We are studying the potential roles of HMGA1a in both transcription and splicing of target genes linked with schizophrenia.
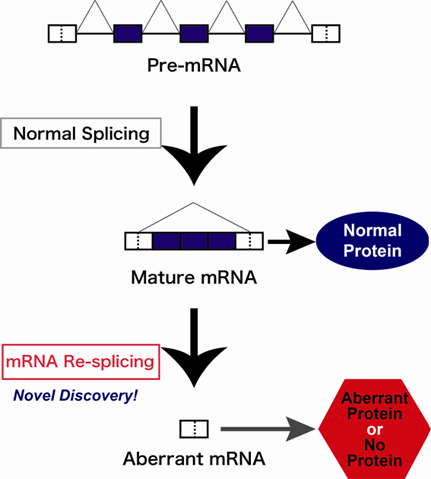
Elucidate the functions of mature mRNA re-splicing discovered in cancer cells
We have recently discovered the cancer-specific aberrant splicing that occurs through re-splicing event in the mature spliced mRNAs of TSG101 and FHIT genes. Usually, in normal cells, spliced mRNA must export to the cytoplasm to serve as a template for the protein biosynthesis. However, such mature mRNA is spliced again and generated aberrant truncated mRNA in cancer cells (see Figure). This finding proposes two important issues to be considered. (i) The mRNA re-splicing events, often induced in cancer cells, implicate the control mechanism to prevent deleterious extra mRNA splicing that must operate in normal cells. (ii) If the mRNA re-splicing is caused by defects in such unknown mRNA quality control mechanism in cancer cells, it could allow the deleterious extra splicing of many mRNAs containing potential splice sites. This scenario is consistent with the fact that aberrant proteins accumulate globally in cancer cells. We are going to perform the extensive analysis to elucidate the proposed issues using cutting-edge high-throughput technique.
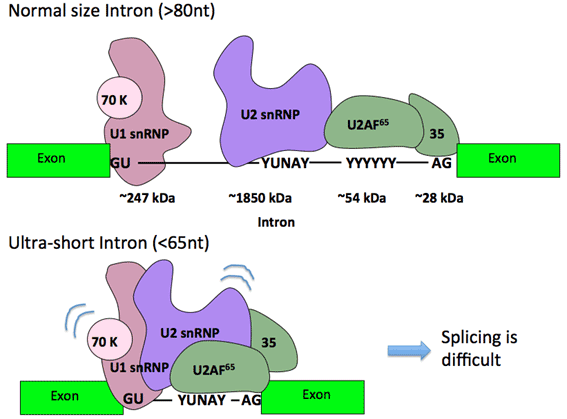
Uncover the splicing mechanism of ultra-short introns in human genes
There are very short introns with <40 nt length in human genome, and actually we do not know about the shortest size of intron. In such an ultra-short introns, the space is evidently too tight to be bound by essential multi-component factors, and it is mysterious about how splicing reaction takes place without steric hindrance. We searched for reliable ultra-short introns shorter than 65 nt with the hallmark GT–AG terminal sequences. We eventually identified 56-, 49-, and 43-nt introns in the HNRNPH1, NDOR1, and ESRP2 genes, respectively, whose splicing were verified in cultured cells. Interestingly, the 49- and 43-nt introns, which lack apparent branch-site sequences, share consensus 11-nt G-rich sequence. We demonstrated that this G-rich sequence functions as an intronic splicing enhancer (ISE), which might be a signal for the recognition of the ultra-short introns lacking branch-site sequences. On the other hand, the splicing of the 56-nt intron requires the exonic sequences in the HNRNPH1. We conclude that the splicing of human ultra-short introns is dependent on splicing enhancer elements in either the intron or the flanking exons. We will extensively analyze the splicing pathway of these ultra-short introns, which may end up in the discovery of a novel mechanism.
Investigate the evolution of splicing factors and the origin of eukaryotic introns
There are several theories about the origin of eukaryotic pre-mRNA introns but we do not know exactly yet. We performed homology search of non-metazoan introns in silico with human spliceosomal proteins as queries. In the case of the microsporidian (Encephalitozoon cuniculi) that has extremely small introns (20–76 nt), half of those spliceosomal proteins are less conserved with the lower score but the conserved proteins has equivalent size with those of metazoan. What is the splicing mechanism of such non-metazoan small introns? We are performing challenging study to shed light on the evolution of splicing factors and origin of introns.