
Unraveling the Molecular Mechanisms Underlying Severe COVID-19
Researchers probe into the detailed molecular pathway which leads to the SARS-CoV-2-induced cytokine storm in severe COVID-19
The levels of proinflammatory cytokines are dramatically elevated in severe COVID-19 patients, the molecular mechanisms of which remain unclear. In a recent article, a group of researchers from Japan has elucidated the role of different molecular players involved in the activation of NF-κB signaling, a key mediator in the cytokine pathway. The findings are quite promising and can lead to the development of potential therapeutic options against COVID-19.
The COVID-19 pandemic has wreaked havoc worldwide. While most infected individuals experience mild symptoms, the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) virus may cause severe pneumonia, acute respiratory distress syndrome, and organ damage in some patients, particularly those with comorbidities. Typically, the symptoms are triggered by an extreme immune response that causes elevated levels of cytokines, referred to as the “cytokine storm.” However, the detailed mechanisms of proinflammatory cytokine production are not fully understood.
Previous studies have reported that the NF-κB family of transcription factors is an integral component of the process. This makes NF-κB signaling an important COVID-related therapeutic target. Interestingly, viral nucleocapsid (N) proteins can also interact with transforming growth factor b-activated kinase 1 (TAK1) and IκB kinase (IKK) complexes to activate the NF-κB pathway. Although there have been speculations to define these interactions, scientists are still trying to determine which viral genes they primarily involve.
To disentangle these mechanisms, a team of researchers, including Professor Takayuki Murata and Dr. Hironiri Nishitisuji from the Fujita Health University School of Medicine in Japan, has conducted a comprehensive study which was recently published in the journal mBio (available online on 20 July 2022). The team, which also contained Dr. Kunitada Shimotohno from the National Centre for Global Health and Medicine, Japan, screened 22 SARS-CoV-2 proteins to identify the ones that could activate NF-κB signaling. In context of the motivation driving the study, Prof. Murata explains, “We tried to identify the modifiers of cell signals, especially those of inflammatory signals, because inflammation is central to the symptoms of COVID-19.”
The team found that two central SARS-CoV-2 proteins—nonstructural protein 6 (NSP6) and open reading frame 7a (ORF7a)—were essential for activating the NF-κB pathway. Consistent with their hypothesis, the expression of NSP6 and ORF7a increased the levels of proinflammatory cytokines such as interleukin 8 (IL-8) and interferon gamma-induced protein 10 (IP-10). This suggests that the viral components may be activating NF-κB to cause the cytokine storm in the later stages of the infection, since infected individuals experience extreme symptoms during severe COVID-19.
Using CRISPR-Cas9 knockout studies, the team further discovered that NSP6 and ORF7a act via transforming growth factor b-activated kinase 1 (TAK1) and NF-κB essential modulator (NEMO), which are crucial players in the NF-κB pathway. Interestingly, the knockdown of TAK1/NEMO significantly reduced SARS-CoV-2-induced NF-κB activation, indicating that these proteins could be considered as potential targets for COVID-19 treatment.
The researchers then looked into the ubiquitination of NSP6 and ORF7a, which could be critical for exerting their downstream effects. Prof. Murata and his team found that tripartite motif-containing 13 (TRIM 13) and ring finger protein 121 (RNF121) were required for the ubiquitination of NSP6 and ORF7a, respectively. This process seemed to be essential for recruiting NEMO to the NSP6-TAK1 complex and activating NF-κB.
Since the suppression of cytokine production and SARS-CoV-2 replication are both halted by blocking NF-κB signaling, it is possible that the virus hijacks the pathway for its replication. “We propose that NF-κB suppression of proinflammatory cytokine responses while maintaining adequate immunity for viral clearance may be a good strategy to stop the virus,” adds Prof. Murata.
Optimistic about the significance of their research, the team anticipates that the study may add to the tool box of existing approaches and treatment ideas for COVID-19. “Our findings offer a better understanding of SARS-CoV-2 pathogenesis and host immune response to the infection”, says Prof. Murata. “Inhibitors of the molecules that mediate NF-κB activation may be used to reduce the severity of COVID-19 symptoms,” he concludes.
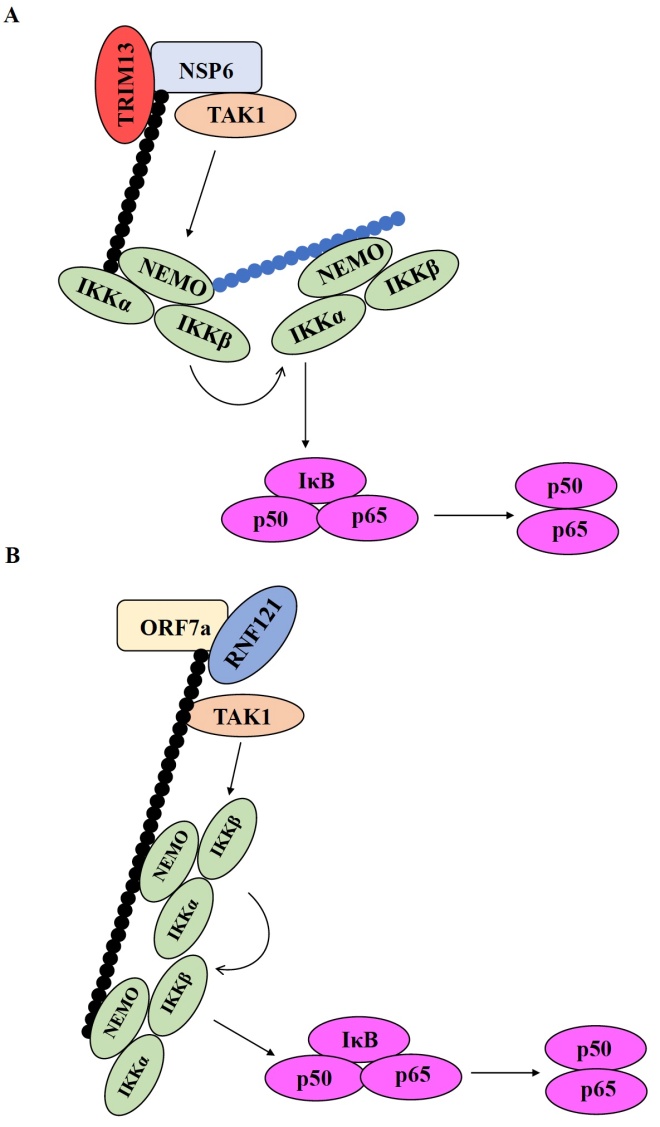
Image title: Possible mechanisms of NF-κB pathway activation in severe COVID-19
Caption: The activation of NF-κB pathway is a complex process where the interaction of different molecular players leads the induction of proinflammatory cytokines. (A) TRIM13 ubiquitinates NSP6, which promotes the recruitment of the IKK/NEMO complex and the activation of NF-κB. Next, NEMO is ubiquitinated, which further promotes the recruitment of the IKK/NEMO complex. (B) RNF121 ubiquitinates ORF7a before recruiting TAK1 and the IKK/NEMO complex to the ubiquitin chain. Ubiquitination of NEMO itself is not needed for NF-κB activation in the case of ORF7a.
Image credit: Prof. Takayuki Murata, Fujita Health University School of Medicine
License type: Original content
The levels of proinflammatory cytokines are dramatically elevated in severe COVID-19 patients, the molecular mechanisms of which remain unclear. In a recent article, a group of researchers from Japan has elucidated the role of different molecular players involved in the activation of NF-κB signaling, a key mediator in the cytokine pathway. The findings are quite promising and can lead to the development of potential therapeutic options against COVID-19.
The COVID-19 pandemic has wreaked havoc worldwide. While most infected individuals experience mild symptoms, the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) virus may cause severe pneumonia, acute respiratory distress syndrome, and organ damage in some patients, particularly those with comorbidities. Typically, the symptoms are triggered by an extreme immune response that causes elevated levels of cytokines, referred to as the “cytokine storm.” However, the detailed mechanisms of proinflammatory cytokine production are not fully understood.
Previous studies have reported that the NF-κB family of transcription factors is an integral component of the process. This makes NF-κB signaling an important COVID-related therapeutic target. Interestingly, viral nucleocapsid (N) proteins can also interact with transforming growth factor b-activated kinase 1 (TAK1) and IκB kinase (IKK) complexes to activate the NF-κB pathway. Although there have been speculations to define these interactions, scientists are still trying to determine which viral genes they primarily involve.
To disentangle these mechanisms, a team of researchers, including Professor Takayuki Murata and Dr. Hironiri Nishitisuji from the Fujita Health University School of Medicine in Japan, has conducted a comprehensive study which was recently published in the journal mBio (available online on 20 July 2022). The team, which also contained Dr. Kunitada Shimotohno from the National Centre for Global Health and Medicine, Japan, screened 22 SARS-CoV-2 proteins to identify the ones that could activate NF-κB signaling. In context of the motivation driving the study, Prof. Murata explains, “We tried to identify the modifiers of cell signals, especially those of inflammatory signals, because inflammation is central to the symptoms of COVID-19.”
The team found that two central SARS-CoV-2 proteins—nonstructural protein 6 (NSP6) and open reading frame 7a (ORF7a)—were essential for activating the NF-κB pathway. Consistent with their hypothesis, the expression of NSP6 and ORF7a increased the levels of proinflammatory cytokines such as interleukin 8 (IL-8) and interferon gamma-induced protein 10 (IP-10). This suggests that the viral components may be activating NF-κB to cause the cytokine storm in the later stages of the infection, since infected individuals experience extreme symptoms during severe COVID-19.
Using CRISPR-Cas9 knockout studies, the team further discovered that NSP6 and ORF7a act via transforming growth factor b-activated kinase 1 (TAK1) and NF-κB essential modulator (NEMO), which are crucial players in the NF-κB pathway. Interestingly, the knockdown of TAK1/NEMO significantly reduced SARS-CoV-2-induced NF-κB activation, indicating that these proteins could be considered as potential targets for COVID-19 treatment.
The researchers then looked into the ubiquitination of NSP6 and ORF7a, which could be critical for exerting their downstream effects. Prof. Murata and his team found that tripartite motif-containing 13 (TRIM 13) and ring finger protein 121 (RNF121) were required for the ubiquitination of NSP6 and ORF7a, respectively. This process seemed to be essential for recruiting NEMO to the NSP6-TAK1 complex and activating NF-κB.
Since the suppression of cytokine production and SARS-CoV-2 replication are both halted by blocking NF-κB signaling, it is possible that the virus hijacks the pathway for its replication. “We propose that NF-κB suppression of proinflammatory cytokine responses while maintaining adequate immunity for viral clearance may be a good strategy to stop the virus,” adds Prof. Murata.
Optimistic about the significance of their research, the team anticipates that the study may add to the tool box of existing approaches and treatment ideas for COVID-19. “Our findings offer a better understanding of SARS-CoV-2 pathogenesis and host immune response to the infection”, says Prof. Murata. “Inhibitors of the molecules that mediate NF-κB activation may be used to reduce the severity of COVID-19 symptoms,” he concludes.
Image title: Possible mechanisms of NF-κB pathway activation in severe COVID-19
Caption: The activation of NF-κB pathway is a complex process where the interaction of different molecular players leads the induction of proinflammatory cytokines. (A) TRIM13 ubiquitinates NSP6, which promotes the recruitment of the IKK/NEMO complex and the activation of NF-κB. Next, NEMO is ubiquitinated, which further promotes the recruitment of the IKK/NEMO complex. (B) RNF121 ubiquitinates ORF7a before recruiting TAK1 and the IKK/NEMO complex to the ubiquitin chain. Ubiquitination of NEMO itself is not needed for NF-κB activation in the case of ORF7a.
Image credit: Prof. Takayuki Murata, Fujita Health University School of Medicine
License type: Original content
Reference
Title of original paper
Ubiquitination of SARS-CoV-2 NSP6 and ORF7a Facilitates NF-κB ActivationJournal
mBioDIO
10.1128/mbio.00971-22About Professor Takayuki Murata
Prof. Takayuki Murata is a professor at the Department of Virology and Parasitology, Fujita Health University School of Medicine, Aichi, Japan. He received his Doctorate in Medical Sciences from Nagoya University. Currently, he is also a visiting professor at Nagoya University, Japan. His major research interests include Virology, Tumor Biology, Epstein-Barr virus, Hepatitis B Virus and SARS-CoV-2. He is a member of the prestigious American Society for Microbiology and the Fujita Medical Society. In his scientific career spanning over two decades, he has more than 100 publications to his credit and has been awarded numerous research grants.
Funding information
This work was supported in part by Takeda Science Foundation