
Novel Method with Implications for Treatment of Fukuyama Muscular Dystrophy—A Widespread Neuromuscular Disorder
Japanese researchers propose using RNA interference for correcting a mistake in the genetic code of patients with Fukuyama Muscular Dystrophy
Fukuyama congenital muscular dystrophy (FCMD) is the second most common form of childhood muscular dystrophy in Japan. The disease manifests itself in the form of a severe neuromuscular disorder. One form of FCMD is caused by a genetic abnormality in the “fukutin” (FKTN) gene. The genetic anomaly blocks the chemical glycosylation of a biologically important protein. Using RNA interference, Japanese researchers restored the normal biological function of FKTN in patient-derived cells, offering hope for therapy.
Muscular dystrophy is a debilitating disease that causes progressive weakening and loss of muscles. Fukuyama congenital muscular dystrophy (FCMD), the second most common form of childhood muscular dystrophy in Japan, is a severe neuromuscular disorder characterized by generalized muscle weakness, decreased muscle tone, eye abnormalities, brain malformation, cardiomyopathy, epilepsy, and seizures associated with intellectual disability.
FCMD is caused by a genetic abnormality in the “fukutin” (FKTN) gene. Researchers from Japan led by Dr. Mariko Taniguchi-Ikeda, an Associate Professor in the Department of Clinical Genetics at Fujita Health University Hospital, were recently able to overcome this defect in the FKTN gene and restore its normal biological function. Using the experimental technique called exon skipping by antisense oligonucleotides the team corrected a mistake in the FKTN gene that blocks the chemical glycosylation of a biologically important protein. To this end, the team designed specific antisense oligonucleotides—small pieces of DNA or RNA that can bind to specific RNA molecules. The researchers then conducted experiments on patient-derived cells using these antisense oligonucleotides to validate their hypothesis.
Dr. Taniguchi-Ikeda elaborates, “I have seen patients with FCMD for more than 20 years. The goal of our research was to find a cure for this intractable disease. FCMD is the second most frequent type of childhood muscular dystrophy in the Japanese population. Our previous studies show that patients who carry retrotransposal insertion can be treated by introducing antisense oligonucleotides. Clinical trials are currently underway. However, no therapeutic methods are available for patients carrying a deep-intronic heterozygous variant. Our findings suggest that exon skipping by antisense oligonucleotides as a treatment for patients with the deep-intronic variant has tremendous potential.”
The basic premise of the research is based on a physiological mechanism involving FKTN. The FKTN gene is responsible for the production of “ribitol-phosphate transferase”—an enzyme that chemically transfers a glycosyl group to alpha-dystroglycan (α-DG). α-DG is a key protein present in the cytoskeleton—a large network of protein filaments and tubules that gives shape and coherence to living cells. The genetic abnormality in the FKTN gene prevents it from expressing a fully functional form of ribitol-phosphate transferase. This reduced functionality, in turn, blocks a critical process in the biological relay—the glycosylation of α-DG. Incidentally, glycosylation, or the attachment of sugar molecules to non-sugar moieties like lipids and proteins, such as α-DG in this case, is important to ensure the structural stability as well as functionality of these moieties.
The findings, which have been published on 25 November 2022 in the journal Human Molecular Genetics, have immediate implications, given the fact that the specifically designed antisense oligonucleotides, when introduced into patient-derived cells via RNAi, were able to skip the affected region of the gene, thus restoring normal production of the FKTN protein and subsequent glycosylation of α-DG.
Radical therapies for certain types of neuromuscular disorders have significantly evolved in recent years. Although no effective therapies were available until recently, several therapeutic approaches have advanced to the clinical stage in the past few years. More specifically, pharmacologic RNA splicing modulation aimed at modifying RNA processing and function has remarkably progressed in recent years.
Co-author Hiroki Kurahashi, a renowned Professor at Fujita Health University adds, “Patients with FCMD who carry the abnormal FKTN gene produce non-glycosylated α-DG, which makes them bedridden right from adolescence. They also require respiratory support, a feeding tube, and lifetime care from their families. Our initial experiments are therefore of paramount significance.”
Encouraged by their in vitro findings, the research team is now proposing translational work by setting up large-scale clinical trials involving patients with FCMD. Lead author Sarantuya Enkhjargal, a Ph.D. student at Fujita Health University Hospital concludes, “The US Food and Drug Administration has approved eight antisense oligonucleotides for the treatment of several diseases. Our findings are promising at the in vitro level. Further studies on the in vivo efficacy and safety in animal models will be needed before this approach is eventually used in clinical trials, however.”
These findings, nonetheless, offer a ray of hope for the multitude of patients and their families affected by this serious condition.
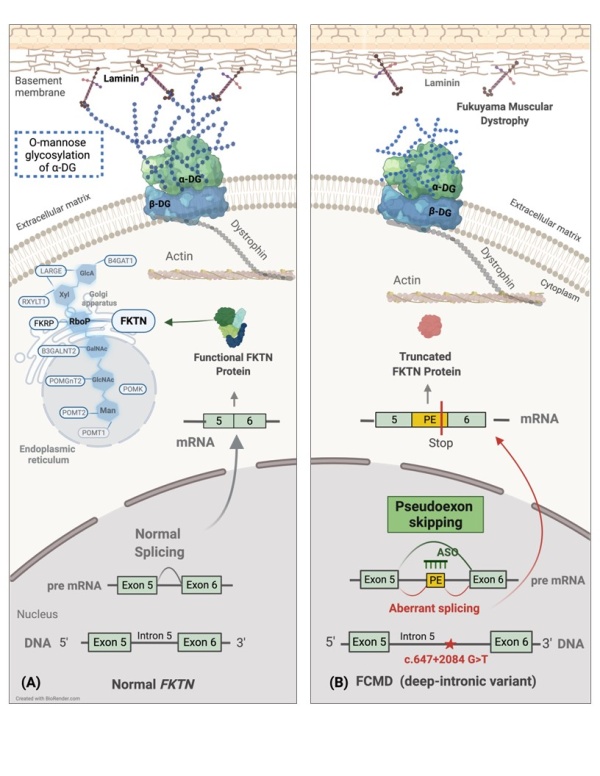
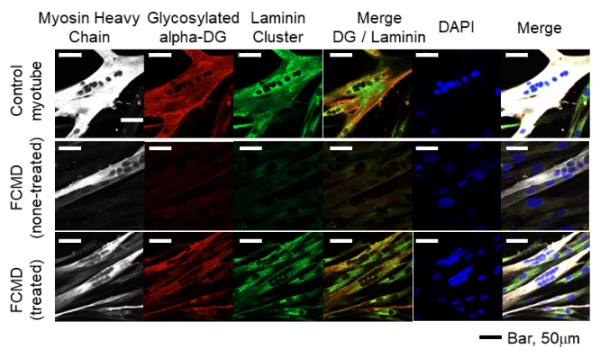
Image title: FKTN production and downstream regulation in untreated and treated patient-derived cells
Image caption: Fluorescent microscopy images showing the absent and restored glycosylated alpha-dystroglycan and function (laminin-binding ability) in patient-derived myotube
Image credit: Mariko Taniguchi-Ikeda from Fujita Health University
License type: Original content
Fukuyama congenital muscular dystrophy (FCMD) is the second most common form of childhood muscular dystrophy in Japan. The disease manifests itself in the form of a severe neuromuscular disorder. One form of FCMD is caused by a genetic abnormality in the “fukutin” (FKTN) gene. The genetic anomaly blocks the chemical glycosylation of a biologically important protein. Using RNA interference, Japanese researchers restored the normal biological function of FKTN in patient-derived cells, offering hope for therapy.
Muscular dystrophy is a debilitating disease that causes progressive weakening and loss of muscles. Fukuyama congenital muscular dystrophy (FCMD), the second most common form of childhood muscular dystrophy in Japan, is a severe neuromuscular disorder characterized by generalized muscle weakness, decreased muscle tone, eye abnormalities, brain malformation, cardiomyopathy, epilepsy, and seizures associated with intellectual disability.
FCMD is caused by a genetic abnormality in the “fukutin” (FKTN) gene. Researchers from Japan led by Dr. Mariko Taniguchi-Ikeda, an Associate Professor in the Department of Clinical Genetics at Fujita Health University Hospital, were recently able to overcome this defect in the FKTN gene and restore its normal biological function. Using the experimental technique called exon skipping by antisense oligonucleotides the team corrected a mistake in the FKTN gene that blocks the chemical glycosylation of a biologically important protein. To this end, the team designed specific antisense oligonucleotides—small pieces of DNA or RNA that can bind to specific RNA molecules. The researchers then conducted experiments on patient-derived cells using these antisense oligonucleotides to validate their hypothesis.
Dr. Taniguchi-Ikeda elaborates, “I have seen patients with FCMD for more than 20 years. The goal of our research was to find a cure for this intractable disease. FCMD is the second most frequent type of childhood muscular dystrophy in the Japanese population. Our previous studies show that patients who carry retrotransposal insertion can be treated by introducing antisense oligonucleotides. Clinical trials are currently underway. However, no therapeutic methods are available for patients carrying a deep-intronic heterozygous variant. Our findings suggest that exon skipping by antisense oligonucleotides as a treatment for patients with the deep-intronic variant has tremendous potential.”
The basic premise of the research is based on a physiological mechanism involving FKTN. The FKTN gene is responsible for the production of “ribitol-phosphate transferase”—an enzyme that chemically transfers a glycosyl group to alpha-dystroglycan (α-DG). α-DG is a key protein present in the cytoskeleton—a large network of protein filaments and tubules that gives shape and coherence to living cells. The genetic abnormality in the FKTN gene prevents it from expressing a fully functional form of ribitol-phosphate transferase. This reduced functionality, in turn, blocks a critical process in the biological relay—the glycosylation of α-DG. Incidentally, glycosylation, or the attachment of sugar molecules to non-sugar moieties like lipids and proteins, such as α-DG in this case, is important to ensure the structural stability as well as functionality of these moieties.
The findings, which have been published on 25 November 2022 in the journal Human Molecular Genetics, have immediate implications, given the fact that the specifically designed antisense oligonucleotides, when introduced into patient-derived cells via RNAi, were able to skip the affected region of the gene, thus restoring normal production of the FKTN protein and subsequent glycosylation of α-DG.
Radical therapies for certain types of neuromuscular disorders have significantly evolved in recent years. Although no effective therapies were available until recently, several therapeutic approaches have advanced to the clinical stage in the past few years. More specifically, pharmacologic RNA splicing modulation aimed at modifying RNA processing and function has remarkably progressed in recent years.
Co-author Hiroki Kurahashi, a renowned Professor at Fujita Health University adds, “Patients with FCMD who carry the abnormal FKTN gene produce non-glycosylated α-DG, which makes them bedridden right from adolescence. They also require respiratory support, a feeding tube, and lifetime care from their families. Our initial experiments are therefore of paramount significance.”
Encouraged by their in vitro findings, the research team is now proposing translational work by setting up large-scale clinical trials involving patients with FCMD. Lead author Sarantuya Enkhjargal, a Ph.D. student at Fujita Health University Hospital concludes, “The US Food and Drug Administration has approved eight antisense oligonucleotides for the treatment of several diseases. Our findings are promising at the in vitro level. Further studies on the in vivo efficacy and safety in animal models will be needed before this approach is eventually used in clinical trials, however.”
These findings, nonetheless, offer a ray of hope for the multitude of patients and their families affected by this serious condition.
Image title: Schematic showing how antisense oligonucleotides restore FKTN protein and α-DG functions in cell-based experiments
Image caption: RNAi-based treatment restores normal FKTN protein production and alpha-dystroglycan glycosylation in cells derived from patients with Fukuyama Muscular Dystrophy carrying the deep intronic variant
Image credit: Mariko Taniguchi-Ikeda from Fujita Health University
License type: Original content
Image caption: RNAi-based treatment restores normal FKTN protein production and alpha-dystroglycan glycosylation in cells derived from patients with Fukuyama Muscular Dystrophy carrying the deep intronic variant
Image credit: Mariko Taniguchi-Ikeda from Fujita Health University
License type: Original content
Image caption: Fluorescent microscopy images showing the absent and restored glycosylated alpha-dystroglycan and function (laminin-binding ability) in patient-derived myotube
Image credit: Mariko Taniguchi-Ikeda from Fujita Health University
License type: Original content
Reference
Title of original paper
Antisense oligonucleotide induced pseudoexon skipping and restoration of functional protein for Fukuyama muscular dystrophy caused by a deep-intronic variantJournal
Human Molecular GeneticsDOI
10.1093/hmg/ddac286About Associate Professor Mariko Taniguchi-Ikeda
Dr. Taniguchi-Ikeda serves as an Associate Professor at the Department of Clinical Genetics, Fujita Health University Hospital. She has over 80 publications and more than 1,300 citations to her credit. Dr. Taniguchi-Ikeda’s research work primarily focuses on Walker-Warburg Syndrome, Fukuyama Type Muscular Dystrophy, Lissencephaly, and X-Linked Retinal Dysplasia. She has published her work in reputed journals such as Nature, PLoS Genetics, and Journal of Human Genetics.
Funding information
This study was supported by grants from the Japan Agency for Medical Research and Development (18ek0109318h0001 to Mariko Taniguchi-Ikeda; 20ek0109405h0002 to Mariko Taniguchi-Ikeda; 18ek0109249h0002 to Tatsushi Toda, Mariko Taniguchi-Ikeda), Grants-in-Aid for Scientific Research (18K07790 to Mariko Taniguchi-Ikeda), The Naito Foundation (to Mariko Taniguchi-Ikeda), Japan Intractable Disease Foundation (to Mariko Taniguchi-Ikeda), Takeda Science Foundation (to Mariko Taniguchi-Ikeda), Houansya Foundation (to Mariko Taniguchi-Ikeda).